I received a vcf generated by DeepVariant + GLnexus. In the FILTER column, there are only two types: . and MONOALLELIC. I was a bit confused about this monoallelic idea.
In the VCF file it was defined as:
##FILTER=<ID=MONOALLELIC,Description=”Site represents one ALT allele in a region with multiple variants that could not be unified into non-overlapping multi-allelic sites”>
This is quite loaded sentence. It is more like a description than a definition.
A better description was given in the GLnexus paper.
Figure 1 gives a very detailed illustration of what it means to be MONOALLELIC.

The caption goes like this:
“Figure 1. Allele unification in joint variant calling. (A) Example abbreviated gVCF records for four participants, giving genome position, reference and alternate alleles, and initial called genotypes. Gray records indicate sequencing coverage for regions with no apparent variation. (B) Schematic view of the alternate alleles seen across the four gVCF inputs; they cluster into two sites except for a spanning deletion allele. (C) Example pVCF representation for these variants, with two multiallelic sites and a third “monoallelic” site representing the deletion allele which could not be unified into the multiallelic sites without introducing phase constraints artificially. The input alleles, genotype calls and (not shown) QC measures from the input gVCF records must be “projected” onto the pVCF site representation.”
To understand this, we first need to understand the g(enome)VCF format. This idea was first introduced in the Isaac paper (Raczy 2013 Bioinformatics) The major difference between a gVCF format and a VCF format is that it not only provides output for all variant, but also the non-variant genomic loci.
This is a bit counter-intuitive. Isn’t that if not specified, we would assume that it is the same as the reference? What further information does it provide?
One apparent distinction is grey lines. “Gray records indicate sequencing coverage for regions with no apparent variation. “ For example the first gray lines in Alice’g gVCF. 22 is chr, 101 is pos, C is REF and the place for ALT is <NR> which stands for <NON_REF> . In the GATK gVCF format doc, <NON_REF> was explained as “This provides us with a way to represent the possibility of having a non-reference allele at this site, and to indicate our confidence either way.” It’s like a placeholder. There is no mutation at this site in this sample, but there might be in other samples a.k.a in this population, and this line holds this place from pos 101 until 101 as specified in END=105.
So there is only one problem, how do we understand the first gray line in Carol’s gVCF. It says END=111, however there is a mutation at 106. This did not happen in the examples given in the GATK gVCF format doc. I checked one of my gVCF files and it also did not happen. Let’s treat it as a typo for now and can come back later when it is relevant. Here let’s look at a chuck of gVCF file with complete records:
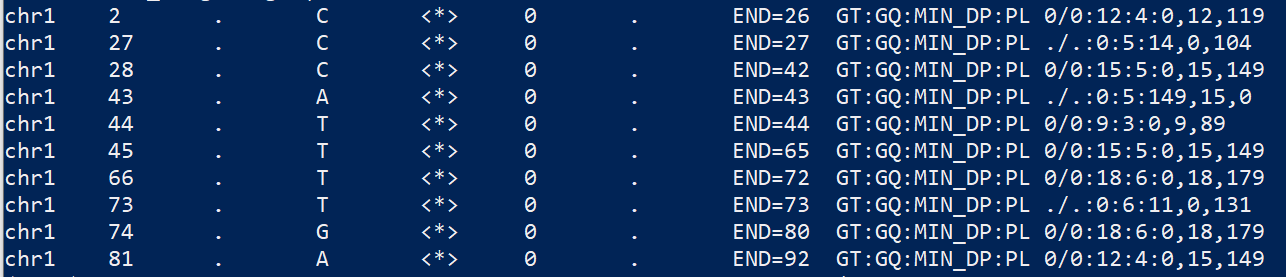
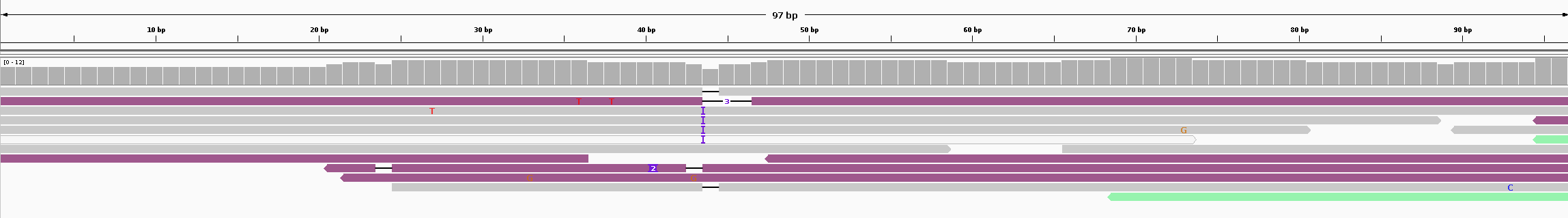
As you can see in the mapping file below, there is no variant called, but any location with possible mutations were document, e.g. line 2, chr1_27 is where the red ‘T’ is.
In my merged pVCF file, there are also MONOALLELLIC sites. One of them is at chr1_10758. It looks like:
chr1 10758 chr1_10758_C_CTA C CTA 37 . AF=0.041252;AQ=37 GT:DP:AD:GQ:PL:RNC 0/0:17:17,0:50:0,51,509:.. chr1 10758 chr1_10758_CCG_C CCG C 30 MONOALLELIC AF=0.029324;AQ=30 GT:DP:AD:GQ:PL:RNC ./.:17:.,0:50:0,0,0:11
Let’s look at the call in the first sample.
We sampled three samples’ gVCFs. In chr1_10758 their call looks like this:
AGO001_08 chr1 10758 . CCG C,<*> 14.7 PASS . GT:GQ:DP:AD:VAF:PL 0/1:5:22:7,15,0:0.681818,0:12,0,3,990,990,990
AGO003_08 chr1 10758 . C CTA,<*> 11.1 PASS . GT:GQ:DP:AD:VAF:PL 1/1:7:10:1,9,0:0.9,0:10,7,0,990,990,990
AGO090_13 chr1 10758 . CCG C,<*> 15.6 PASS . GT:GQ:DP:AD:VAF:PL 1/1:10:10:1,9,0:0.9,0:15,10,0,990,990,990
In the merged pVCF their genetypes are:
./0:22:7,0:3:0,0,0:-. 1/1:10:1,9:1:10,7,0:.. ./.:10:1,0:6:0,0,0:– ./1:22:.,15:5:0,0,0:1. ./.:10:.,0:3:0,0,0:11 1/1:10:.,9:3:0,0,0:..
Some explaination: e.g ./0:22:7,0:3:0,0,0:-.
- GT: genotype. ./0 is a half call. It means there are enough evidence for Ref but not enought to make it a 0/0 call (7 is not big enough).
- DP: read depth 22
- AD: Allelic depths for the ref and alt alleles in the order listed 7,0.
- GQ: conditional Genotype Quality, encoded as a phred quality -10logP (genotype call is wrong, conditioned on the site’s being variant). The higher the better. 3 is pretty low.
- PL: the Phred-scaled genotype Likelihoods rounded to the closest integer. The lower the better. 0,0,0. All equally low.
- RNC: Description=”Reason for No Call in GT: . = n/a, M = Missing data, P = Partial data, I = gVCF input site is non-called, D = insufficient Depth of coverage, - = unrepresentable overlapping deletion, L = Lost/unrepresentable allele (other than deletion), U = multiple Unphased variants present, O = multiple Overlapping variants present, 1 = site is Monoallelic, no assertion about presence of REF or ALT allele”> In our example there are five types of RNC in those 6 calls:
- ’–’: both phase “unrepresentable overlapping deletion”.
- ’..’: n/a (there are calls, therefore no “Reason for No CAll”)
- ’-.’: there is one call (n/a), the other half is due to “unrepresentable overlapping deletion”
- ‘1.’: there is one call (n/a), the other half is due to “site is Monoallelic, no assertion about presence of REF or ALT allele”
- ‘11’: “site is Monoallelic, no assertion about presence of REF or ALT allele”
OK so in this case, the insertion called in AGO003_08 wasn’t included in the merged call. Maybe it was only found in this sample. 9 Reads, actually not bad. Both AGO001_08 and AGO090_13 had the same call: CCG_C, while the former had a R/A call (0/1) and the latter had a A/A call. Why? The former nad 7 reads for R and 15 reads for A, while the later had 1 read for R and 9 reads for A. Fair enough.
In sum, a MONOALLELIC row in a pVCF file generated by GLnexus is a result of inability to merge all called genotypes at the same site into a single multiallelic variant. In the case of position 105 on chromosome 22 in Figure 1, they were able to do it by extending int position 106 and 107. But in position 100, we cannot extend chr22_100_T_C to 105 to accommodate chr22_100_TCGTCA_T, otherwise you would be merging an indel with a SNV. By the way I just learnt that SNP is a smaller concept than SNV. Only SNV found in >1% of the population can be called SNP.
Apparently this is not a new concept. In a 1995 Nature Genetics paper, Papadopoulos et. al used it in the most literal way: Monoallelic mutations are “mutations (that) occur in only one allele”, which could be represented as 0/1 in a diploid genome (please note that since I work on a diploid genome so everything I wrote on my website would be under this assumption unless specified otherwise). While here it is referring to that the REF part of the call in this position is already shown in the variant above and here they are only showing the presence of ALT, thus the mono.
I checked the same position called via freebayes. It combined it with an adjacent variant (chr1_10760) into a multiallelic site. It looks like:
chr1_10758 CCGTAG CTACGG,CCATAG
I would say I prefer this version, so I do not need to deal with the MONOALLELIC situation where I would have filtered it out due to the high presence of “missed calls”. But I would love filtered out this version called by freebayes as well since it is not bi-allelic. What would you do? Leave me a comment.